
Dr. Monica Fernández-Quintero, University of Innsbruck, Austria
Antibody’s Next Top Model – Use of CDR loop ensembles from molecular dynamics simulations guides antibody design and docking
The demand for precise antibody structure models has led to a new paradigm in the understanding of antibody paratopes as ensembles in solution. Characterizing an antibody with only a single-static structure limits the understanding of its function, whereas knowledge of various biophysical properties governed by its dynamic character, e.g., antibody-antigen binding, enables improved antibody design.
The immune system comprises numerous sophisticated processes and mechanisms to protect the body. Central to various defense mechanisms are antibodies, which represent a unique class of proteins with regard to their structural and chemical properties. Their ability to recognize and bind different antigens with high specificity, together with their modular anatomy, makes antibodies excellent therapeutic proteins and facilitates their engineering and design. [1] However, structure-based biophysical determinants for their binding specificity and affinity must be understood before suitable antibody therapeutics can be designed. The structural characterization of antibody-antigen interactions has provided valuable insights into their binding mechanism. The increase of experimentally available antibody structures has enabled numerous high-quality structure-function relationship studies, which have helped to elucidate antibody characteristics and different antigen binding mechanisms. These structure-function relationship studies have created a demand for precise homology models of antibody structures, which allow rational antibody design and engineering when no crystal structures are available. Even though the number of experimental structures has increased substantially, the vast size of immune repertoires and the countless number of antigens make it impossible to structurally and functionally characterize all antibodies and antibody-antigen complexes.
To meet this challenge, numerous computational approaches that aim at improving antibody structure prediction and the prediction of antibody-antigen complexes and their binding affinities have been developed. [2] One of these methods is docking, which attempts to predict antibody-antigen complexes, when no experimentally determined structure is available. [3] By using the individual proteins as starting points, docking generates thousands of possible interaction poses, among which the biological relevant one must identified. Despite various advances in the field, protein-protein docking still remains challenging. The fact, that proteins, in particular antibodies, are highly flexible and can undergo substantial conformational rearrangements upon binding limits the accuracy of the final docking outcome. The flexibility of antibodies is concentrated on one specific region of the antibody, the antigen-binding fragment (Fab), which plays a major role in antigen-recognition and binding. Thus, the three-dimensional structure of the antibody strongly determines its function and binding properties. The Fab consists of a heavy and a light chain and can be divided into one constant (CH-CL) and one variable domain (VH-VL). The highest diversity of the Fab is located in the variable fragment (Fv) and concentrated on six hypervariable loops, also known as the complementarity-determining regions (CDRs). Together with the CDR loops, the relative interdomain orientation VH-VL co-determines the shape of the antigen-binding site, the paratope. In agreement with NMR, our group could show that even short molecular dynamics simulations are sufficient to capture variations up to 30° in the interface (VH-VL, CH1-CL) and elbow angle dynamics in Fabs, as they occur in the low nanosecond timescale. [4,5]
While the framework of the Fv is structurally highly conserved, structure prediction of the relative interdomain orientations and the CDR loops remain challenging. To facilitate antibody structure prediction, five of the six CDR loops (CDR-L1, CDR-L2, CDR-L3, CDR-H1 and CDR-H2 loops) have been classified into so-called canonical clusters, assuming that these loops can only adopt a limited number of distinct backbone conformations. The canonical structure assignment is based on the length and sequence of the respective CDR loop. Because of the high diversity in length, sequence and structure of the CDR-H3 loop, no canonical cluster structure can be assigned. The CDR-H3 loop is located at the center of the paratope, and thereby plays a key role in molecular recognition. Additionally, the length and structure of the CDR-H3 loop can directly alter antigen-binding patterns, and thereby directly affect the specificity of the paratope. Thus, structurally characterizing the CDR-H3 loop and the influence of conformational rearrangements on other CDR loops and the relative VH-VL orientation is a key step in understanding antibody structure and function.
Apart from the CDR loops, another critical aspect in antibody design is the antibody framework. Already single-point mutations in the framework can result in different CDR loop conformations in solution, and thereby directly influence properties of the paratope, such as specificity, stability, and hydrophobicity. In line with these observations, it has been shown that ~ 22% of the residues that interact with the antigen fall outside of the traditionally defined CDR loops. [6] This result raises the question: Do single static canonical structures account for these correlated loop and interface rearrangements as a consequence of affinity maturation, humanization, different heavy and light chain pairings and/or single-point mutations in the antibody framework (e.g., vernier zone residues)?
The long-believed paradigm of static canonical structures determining binding properties and specificity of antibodies has recently shifted, based on our studies. [7,8] We revealed that one single static structure is not sufficient to capture the high flexibility of all CDR loops, in particular of the CDR-H3 loop, and thus the paratope should be described as conformational ensemble in solution. In contrast to this prevalent static view of the binding interface, we demonstrate a dynamic perspective not only of the paratope, but of whole Fvs and Fabs. We show that antibodies exist as ensembles of paratope states. These paratope states are defined by a characteristic combination of CDR loop conformations and interdomain orientations, which interconvert into each other in the micro-to-millisecond timescale by correlated loop and interdomain rearrangements. Thus, to fully understand the antibody binding interface and to elucidate the molecular mechanism of antibody-antigen binding, the strongly correlated CDR loop conformations and the interfaces should be characterized as ensembles in solution.
This dynamic view of the antibody binding site is particularly important to identify dominant conformations in solution when the available X-ray structure is distorted by crystal packing effects or if no experimental structure has been determined. In various examples, we have observed for the CDR-H3 loop and for the whole antibody paratope that the dominant ensemble in solution coincides with the binding competent conformation, which is not necessarily the case with the (apo) X-ray structure.
This high conformational diversity of the binding site is a special feature of antibodies, which allows them to recognize and neutralize more than one antigen. This theory was already suggested by Pauling and Landsteiner, who proposed that the antigen selects a conformation from the pre-existing antibody conformations in solution. [9,10] This ensemble view has been supported by the conformational selection model, which assumes that antibodies exchange between conformational states with varying probabilities. Upon binding, these populations will shift towards the binding competent conformation.
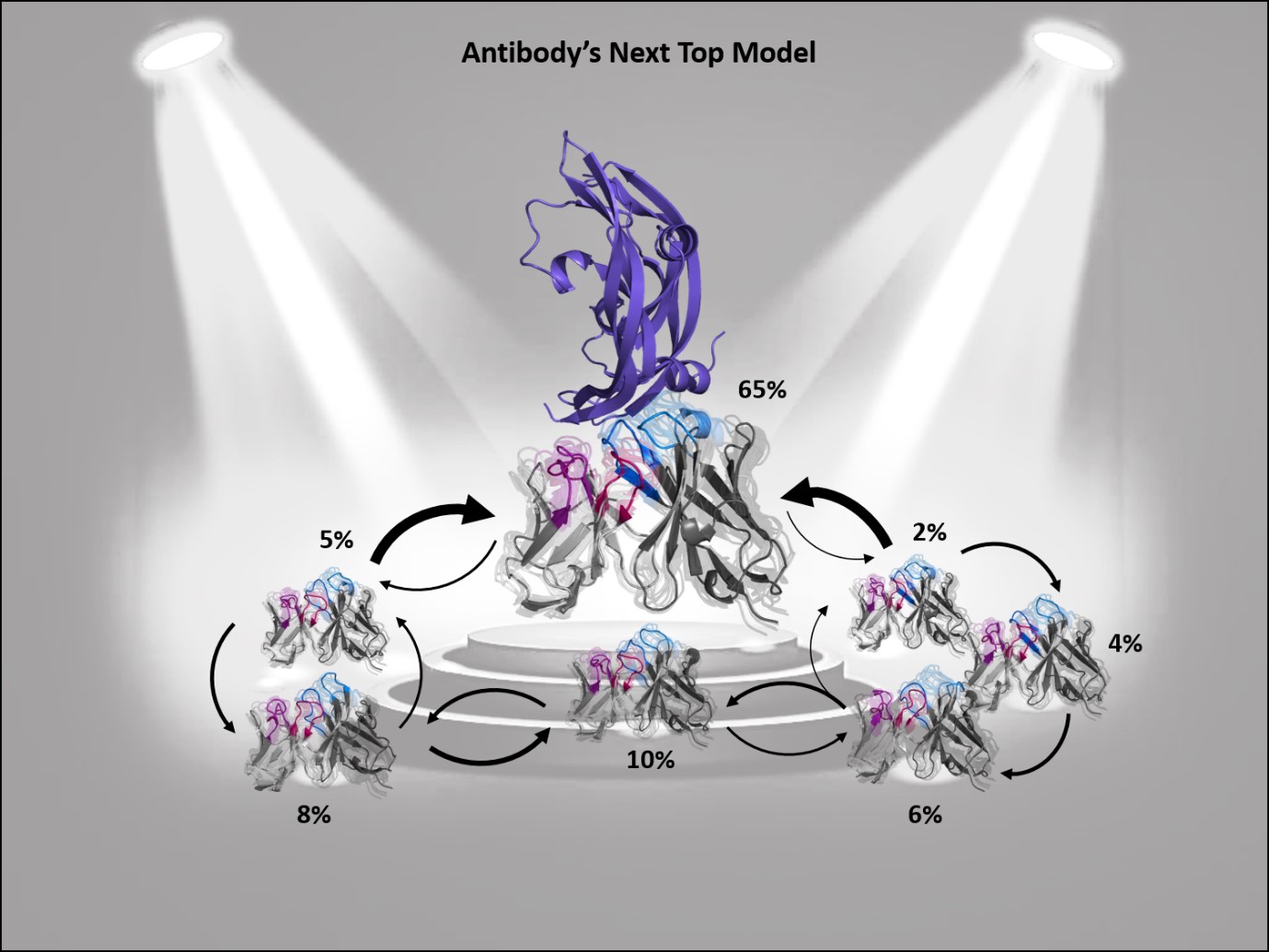
Figure 1: Ensembles in solution improve antibody structure prediction and docking. Different interconverting paratope ensembles in solution with varying probabilities and transitions timescales. The dominant ensemble in solution is in the spotlight, as the binding competent conformation often coincides with the highest populated state.
Consequently, to predict antibody-complexes with docking, the high flexibility of the paratope by considering multiple CDR loop conformations, often referred to as ensemble docking or flexible docking, must be taken into consideration. However, incorporating multiple paratope conformations only increases accuracy when all important ensembles in solution are sampled and thermodynamically weighted by their respective ensemble probabilities (Figure 1). Otherwise, the inclusion of distinct paratope conformations without thermodynamic or kinetic information might introduce more inaccuracies and noise to the docking results. Thus, to benefit from paratope ensembles in solution, both structural and thermodynamic information is critical, as without such thermodynamic weighting, high energy conformations could dominate the docking results and consequently deteriorate the docking success.
In summary, describing an antibody’s binding site using only one single static structure limits the understanding and characterization of the antibody’s function and properties. This limitation is even more pronounced when no experimentally determined structure is available or the crystal structure is distorted by packing effects, which can result in misleading antibody paratope structures. To improve antibody structure prediction and to take the strongly correlated CDR loop and interface movements into account, antibody paratopes should be described as interconverting states in solution with varying probabilities. These kinetically characterized paratope ensembles with their respective state probabilities allow the identification of the dominant conformation in solution, which frequently has been shown to coincide with the binding competent conformation. Therefore, the definition of kinetically and functionally relevant states, so-called paratope states, can be successfully used to improve the accuracy and enhance the predictivity of antibody-antigen docking.
References
1. Chiu ML, Goulet DR, Teplyakov A, Gilliland GL. Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies (Basel) 2019; 8:55.
2. Guest JD, Vreven T, Zhou J, Moal I, Jeliazkov JR, Gray JJ, Weng Z, Pierce BG. An expanded benchmark for antibody-antigen docking and affinity prediction reveals insights into antibody recognition determinants. Structure 2021; 29(6):606-621.
3. Chaudhury S, Gray JJ. Conformer Selection and Induced Fit in Flexible Backbone Protein–Protein Docking Using Computational and NMR Ensembles. Journal of Molecular Biology 2008; 381:1068–87.
4. Fernández-Quintero ML, Kroell KB, Heiss MC, Loeffler JR, Quoika PK, Waibl F, Bujotzek A, Moessner E, Georges G, Liedl KR. Surprisingly Fast Interface and Elbow Angle Dynamics of Antigen-Binding Fragments. Frontiers in Molecular Biosciences 2020; 7:339.
5. Fernández-Quintero ML, Hoerschinger VJ, Lamp LM, Bujotzek A, Georges G, Liedl KR. VH-VL interdomain dynamics observed by computer simulations and NMR. Proteins: Structure, Function, and Bioinformatics 2020; 88(7):830-839.
6. Kunik V, Ashkenazi S, Ofran Y. Paratome: an online tool for systematic identification of antigen-binding regions in antibodies based on sequence or structure. Nucleic Acids Research 2012; 40:521–4.
7. Fernández-Quintero ML, Kroell KB, Hofer F, Riccabona JR, Liedl KR. Mutation of Framework Residue H71 Results in Different Antibody Paratope States in Solution. Frontiers in Immunology 2021; 12:243.
8. Fernández-Quintero ML, Pomarici ND, Math BA, Kroell KB, Waibl F, Bujotzek A, Georges G, Liedl KR. Antibodies exhibit multiple paratope states influencing VH–VL domain orientations. Communications Biology 2020; 3:589.
9. Landsteiner Karl. The specificity of serological reactions. Journal of Pharmaceutical Sciences 1963; 52:1196–1196.
10. Pauling L. A Theory of the Structure and Process of Formation of Antibodies*. Journal of the American Chemical Society 1940; 62:2643-57.