
 Registration is open for our next webinar, “Modeling Biologic Molecules“, to be held Thursday January 27, 2022, 11am ET / 5pm CET.
Registration is open for our next webinar, “Modeling Biologic Molecules“, to be held Thursday January 27, 2022, 11am ET / 5pm CET.
The speakers are Dr. Monica Fernández-Quintero and Prof. Klaus R. Liedl.
Modeling in Chemistry obviously depends on a strong link to reality. Even though the mathematical description of chemistry has been possible for almost 100 years, realistic modelling has only recently become available due to the recent massive increase of computing power following Moore’s law. Still, appropriate statistics, initial conditions and boundaries pose considerable challenges. Nowadays, methodological advances and progress in hardware allows the observation of biological systems for relevant time periods. Hence, dynamic processes like reorientations, folding and binding can be seen in atomistic resolution leading to completely new insights.
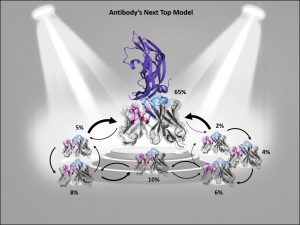
Describing an antibody’s binding site using only one single static structure limits the understanding and characterization of the antibody’s function and properties, whereas various biophysical properties are governed by its dynamics, e.g., antibody-antigen binding. This limitation is even more pronounced when no experimentally determined structure is available or the crystal structure is distorted by packing effects, which can result in misleading antibody paratope structures. To improve antibody structure prediction and to take the strongly correlated CDR loop and interface movements into account, antibody paratopes should be described as interconverting states in solution with varying probabilities. These kinetically characterized paratope ensembles with their respective state probabilities allow the identification of the dominant conformation in solution, which frequently has been shown to coincide with the binding competent conformation. Therefore, the definition of kinetically and functionally relevant states, so-called paratope states, can be successfully used to improve the accuracy and enhance the predictivity of antibody-antigen docking.